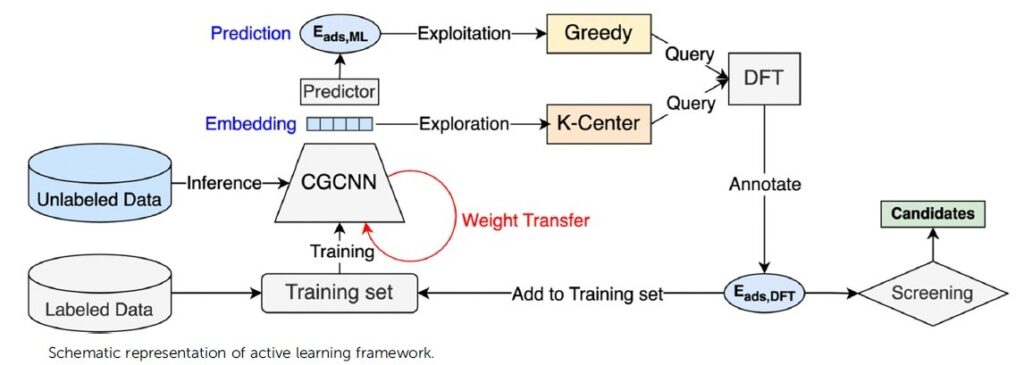
In this research area, we contribute to the development of new materials, in particular porous materials, for efficient carbon capture. Integrated with the experiment, our theoretical analysis provides a mechanistic understanding on the adsorption properties ranging from the thermodynamic limit such as adsorption uptake to the kinetic event such as diffusion process. Our interest is not limited to the mechanism analysis. The high-throughput simulation is one of the key advantages of in-silico approaches, providing the efficient pathway to the material discovering which cannot be easily accessed via the experimental screening.
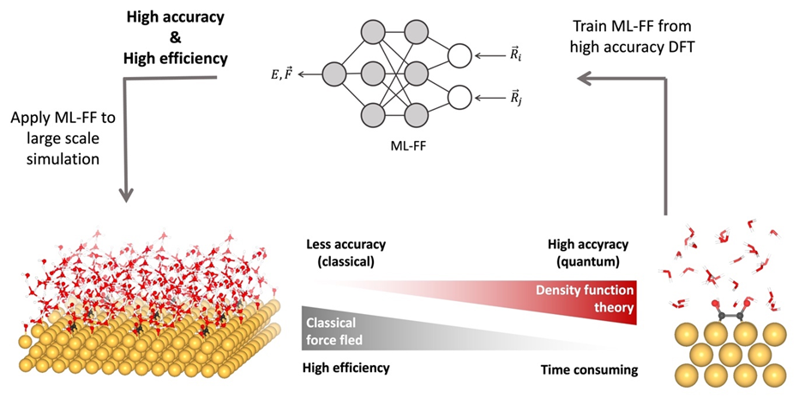
The molecular dynamics (MD) and the grand canonical Monte Carlo (GCMC) based on classical force field are used in our study. For high-precision prediction, we integrate the machine learning potential (MLP) trained at quantum mechanics level to MD simulations, rendering high prediction ability surpassing the conventional classical force field.
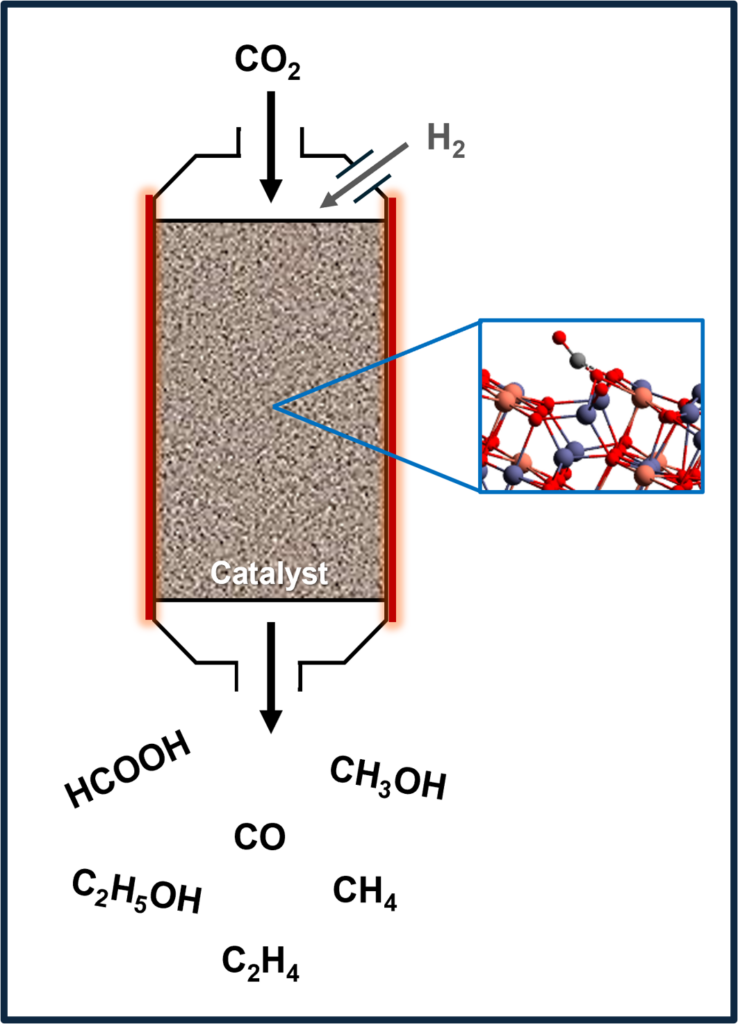
Carbon dioxide utilization: Thermocatalysis
Thermal catalysis is the preferred method for carbon utilization on an industrial scale due to its straightforward process control and the reusability of catalytic materials. Despite these advantages, the high reaction temperatures required usually result in many side reactions and easy catalyst degradation, thereby reducing overall process efficiency. To overcome these limitations, our team employs advanced computational techniques such as Density Functional Theory (DFT) to enable the rational design of novel, high-performance catalytic materials and optimize existing catalysts. The research focuses on identifying active sites, elucidating reaction mechanisms, improving conversion efficiency, enhancing product selectivity, and minimizing thermal energy consumption. Through these efforts, we aim to develop more efficient catalytic processes that provide both economic and environmental benefits by converting CO₂ into value-added chemicals.
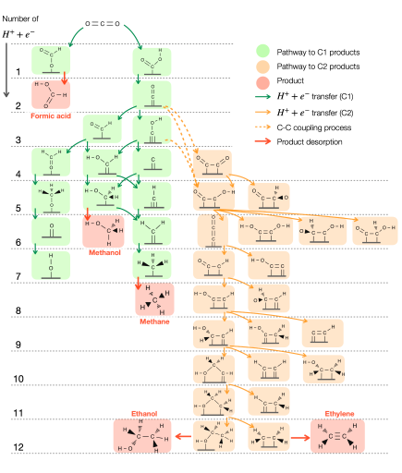
Carbon dioxide utilization: Electrocatalysis
Proposed reaction mechanism of CO2 electroreduction to C1 and C2 products
We focus on developing catalysts for CO2 conversion into valuable chemicals, ranging from C1 to C2 products, including urea synthesis. By leveraging in-silico simulations through multiscale modeling combined with machine learning, we efficiently screen materials for high catalytic performance. Our research also investigates solvent effects at electrode interfaces, applied potentials, and the electronic properties of catalysts, offering atomic-scale insights into reaction mechanisms and interfacial phenomena.
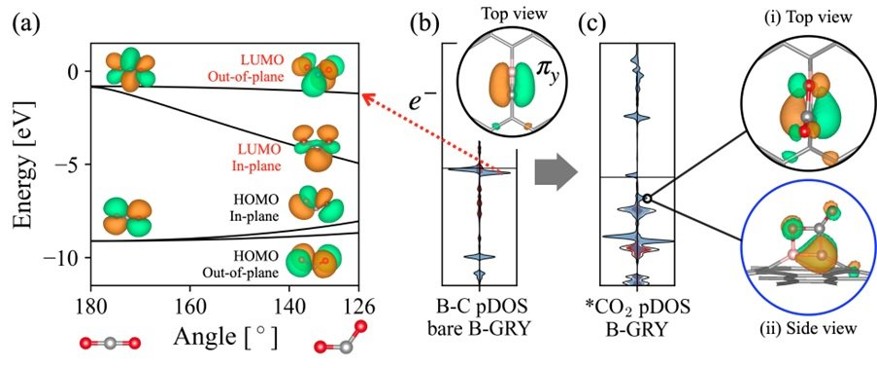
Analysis of CO2 adsorption on Boron doped graphyne
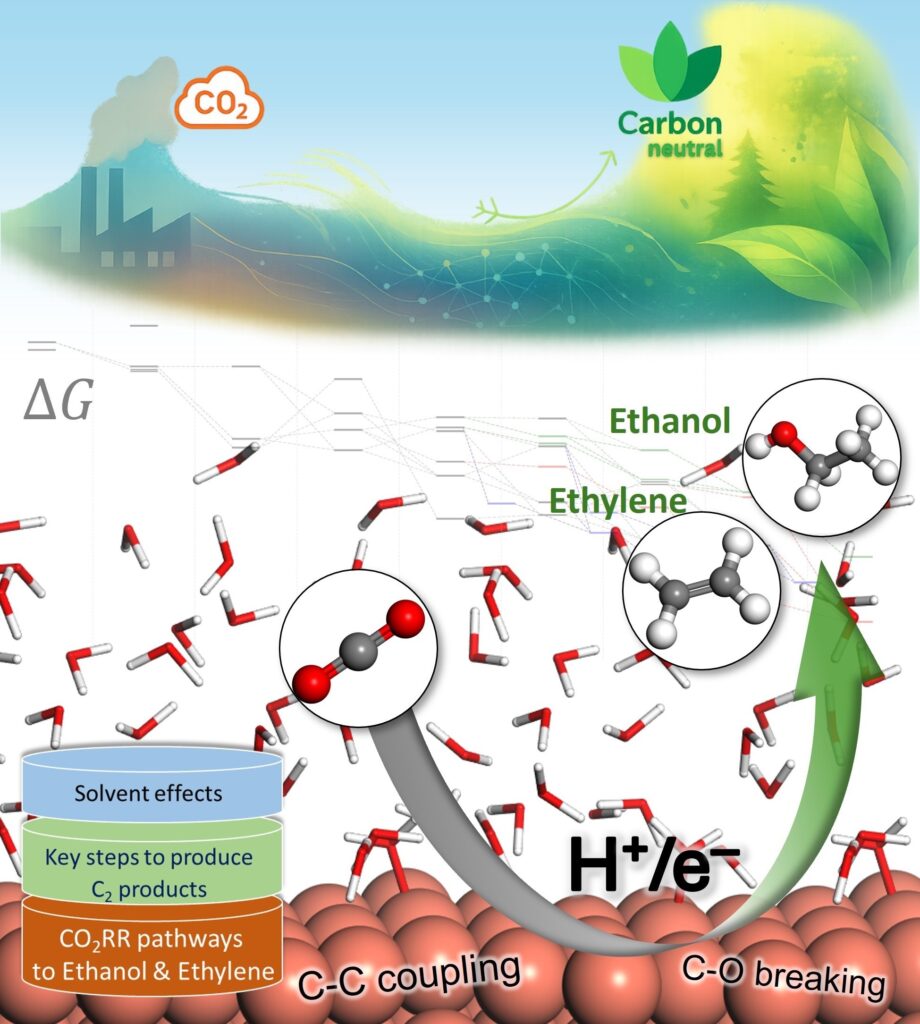
To deepen our understanding of electrocatalysts for carbon utilization, we investigate the atomic-level mechanisms of CO2 electroreduction on Cu-based catalysts. Particular focus is given to reaction pathways leading to multicarbon products, using DFT calculations on interface models constructed with explicit water layers. Interfacial solvent structures are equilibrated via ab initio molecular dynamics (AIMD) simulations to reflect representative local environments near the catalyst surface.
This framework allows key reaction steps, such as CO dimerization, proton–electron transfers, and C–O bond cleavage, and key intermediates to be examined in the presence of solvent. The approach provides mechanistic insight into how solvation influences intermediate stability and product branching between ethylene and ethanol. Beyond individual pathways, this methodology supports the identification of selectivity descriptors and offers a transferable platform for understanding and improving Cu-based electrocatalysts in CO2-to-fuel conversion, helping bridge fundamental insight with practical catalyst design.
Carbon dioxide utilization: Photocatalysis
Our research focuses on photocatalysis, utilizing Metal-Organic Frameworks (MOFs) to efficiently convert CO2 into valuable C1 and C2 products. We investigate how variations in MOF structure and composition influence catalytic activity and product selectivity. By employing advanced DFT calculations, we analyze electronic structure, charge transfer, and reaction mechanisms. These insights support the rational design of MOFs for efficient solar-to-chemical energy conversion.
Materials for pollutant removal
Porous Materials for Clean Water
We are engineering novel porous materials, especially MOFs, for the efficient removal of harmful water pollutants like fluoride (F⁻) and per- and polyfluoroalkyl substances (PFAS). Our research employs DFT and Molecular Dynamics (MD) simulations to deeply understand pollutant adsorption and dynamics. These computational findings are crucial for developing highly selective and effective solutions for water purification and environmental protection.
Adsorbents and Catalysts for Clean Air
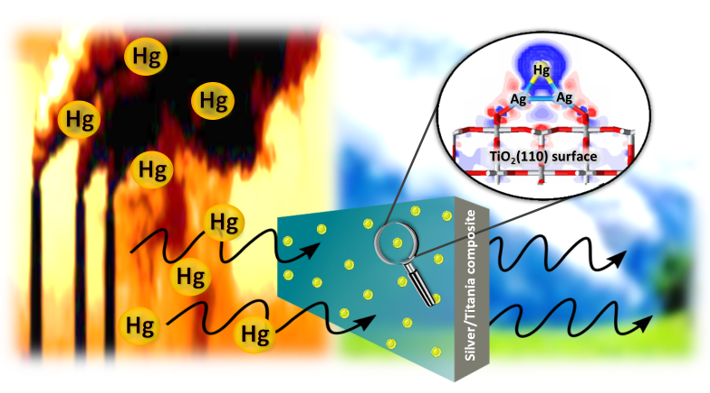
We focus on the computational design and analysis of materials for air pollutant removal, targeting contaminants such as vapor-phase mercury, NOx, SOx, volatile organic compounds (VOCs), and PM2.5 precursors. By applying theoretical methods, we aim to uncover the fundamental mechanisms governing adsorption, surface reactions, and pollutant decomposition. These insights help guide the development of advanced adsorbents and catalysts with enhanced performance for clean air applications.
Catalysis for high-value chemical production
Our research aims to advance the rational design of heterogeneous catalysts for the efficient and selective production of high-value chemicals. We employ state-of-the-art computational methods—such as density functional theory (DFT), molecular dynamics (MD), and microkinetic modeling—to investigate surface reactions, active site structures, and reaction pathways at the atomic level. Simulations, when supported by experimental evidence, help translate theoretical insights into practical catalytic solutions.
Through first-principles simulations, we gain mechanistic insights into complex catalytic processes, enabling the identification of key descriptors that govern activity, selectivity, and stability. These insights are critical for developing next-generation catalysts for challenging chemical transformations such as methane activation, biofuel and biochemical production, dehydrogenation and hydrogenation, selective oxidation, and other reactions.
Materials for energy and devices
Metal-ion batteries
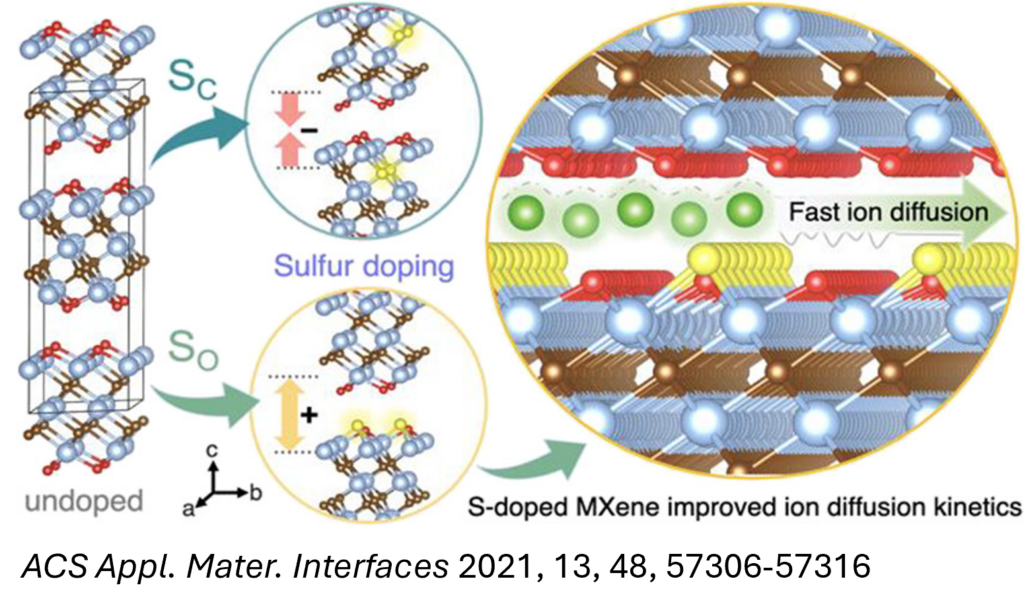
Computational methods, including DFT, AIMD, and kMC simulations, have been employed to investigate and design electrode materials for alkali-ion batteries, such as Li-ion, Na-ion, and Mg-ion systems. Insights gained from key calculated properties—such as electronic conductivity, ion intercalation kinetics, and structural stability—provide a robust framework for enhancing the electrochemical performance of electrode materials. We employ these simulation techniques to explore transition metal carbides (MXenes) and their heterostructures, which show considerable promise for high-performance energy storage applications.
Solar cell and Organic light-emitting diode (OLEDs)
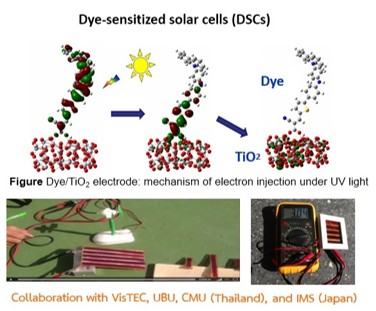
The performance of solar cells and OLEDs strongly depends on the geometry, electronic structure, and excited-state properties of the active materials. In our research, we employ density functional theory (DFT), time-dependent DFT (TD-DFT), and the symmetry-adapted cluster-configuration interaction (SAC-CI) methods to investigate these properties and provide theoretical insights that complement and explain experimental findings. Our combined theoretical and experimental studies highlight key factors that enhance device performance, including efficient charge transfer, high molar extinction coefficients, and reduced molecular aggregation.
Gas sensor
This research focuses on developing gas sensors such as for VOCs, CO particularly for nitrogen-containing gases (e.g., NH3, NO, NO2) using density functional theory (DFT) and machine learning (ML). These gases, often released from nitrogen-based fertilizers, are key contributors to PM2.5 air pollution. We use DFT to study gas adsorption and electronic changes in sensor materials, providing atomic-level insight into sensing mechanisms. ML models trained on DFT data enable rapid screening of materials for high sensitivity and selectivity. Our integrated approach supports the design of advanced, low-cost sensors for real-time monitoring of nitrogenous emissions, contributing to air quality management and pollution control.
Simulation tools development
Machine Learning Potential (MLP)
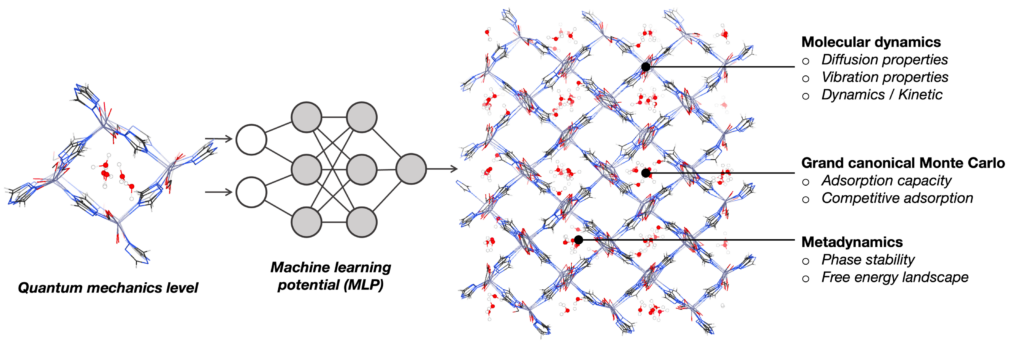
Although ab initio quantum chemistry calculations have been providing a great impact on fundamental understanding of materials, the applicable model size is limited to several hundred atoms within femto-to-picosecond of timescale. In our team, we utilize MLP trained at DFT level to extend the applicability in both length scale (up to several-thousand atoms) and time scale (up to nanoseconds) to enhance the thermodynamics sampling at quantum chemistry level. This approach gives an access to the key information connected to the experimental observables, for example, gas diffusion property inside materials, the local structure and lattice fluctuation, phase stability, and the bond forming-breaking as a function of time and the theoretical prediction of the infrared (IR) spectra.
Post-processing of the molecular dynamics
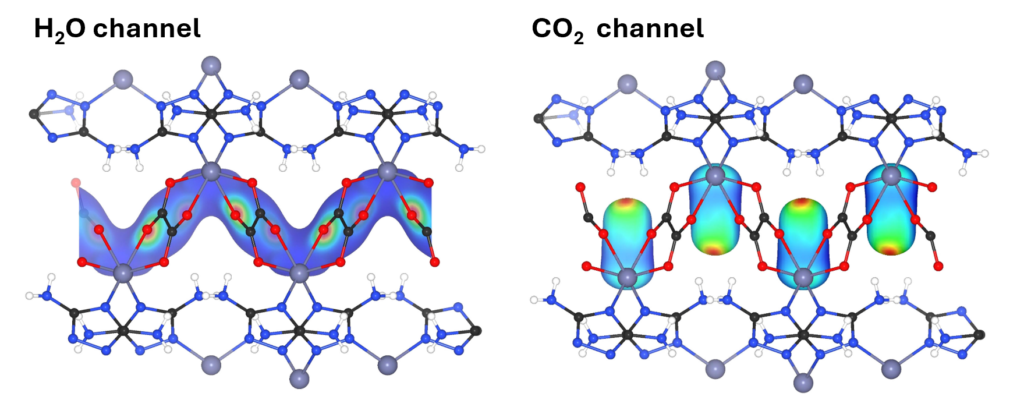
The great impact of molecular dynamics simulation can be achieved by post-processing. We develop in-house code for the calculation, for example, the atomic density distribution analysis providing the information of key binding sites in porous materials, the autocorrelation function for measuring the correlation time of physical properties and the infrared spectra calculation providing the vibrational properties as a fingerprint of materials.
Realistic electrode/electrolyte interface models
Development of realistic electrode/electrolyte interface reactions using MLPs
Electrochemical reactions occur at the electrode-electrolyte interface, where the electrolyte interacts with the electrode under applied potentials. To understand this complex environment, we are developing computational tools that model the catalyst surface, liquid electrolyte, and applied potential using multiscale approaches and machine learning potentials (MLPs). These tools accelerate molecular dynamics (MD) simulations, enabling larger systems and longer time scales. Beyond electrochemical energy conversion, the tools are applicable to a wide range of solid-liquid interface systems, including batteries, sensors, corrosion, and photoconversion. By combining atomistic simulations with experimental validation, we aim to bridge theory and experiment, advancing realistic electrochemical modeling for carbon neutrality and sustainable energy.
This website uses cookies to offer you a seamless experience. These cookies are essential for running our website and are key to providing you a smoother and more personalized experience. By using our website, you acknowledge that you have read and understand our cookie policy.