National Science and Technology Development Agency | National Nanotechology Center
Research
Molecular Simulation
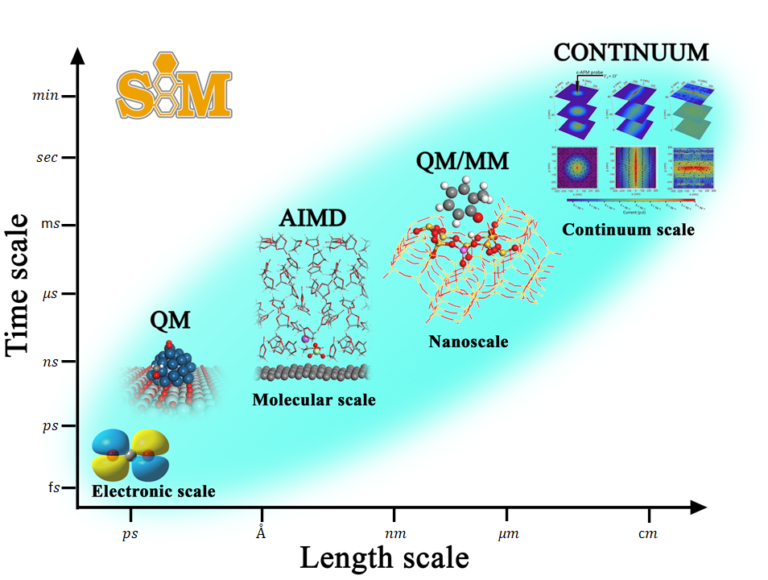
Nanoscale simulation research team (SIM) has extensive experiences in the nanoscale modeling and simulations for both fundamental scientific findings and practical industrial significances. We aim to design and predict the properties of functional nanomaterials, and to create a unique understanding of the physicochemical processes at the nanoscale level. Our expertise includes first-principles computational approaches, molecular dynamics simulation, and hybrid QM/MM simulation, enabling the in-depth analysis of molecular interactions and electronic transports for various applications such as catalysis, pollutant removal, and energy devices. This ‘in silico’ insight can accelerate the experimental research and development of nanotechnology applications in the fields of energy and environment.
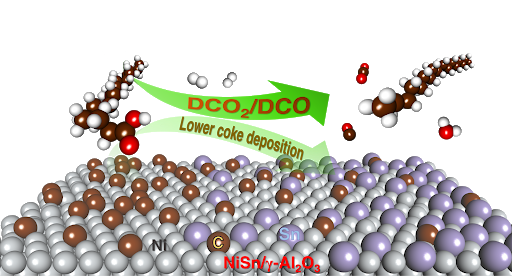
CATALYSIS FOR BIOREFINERY
Efficient catalytic systems, which require less reactions, separation and purification processes, have been sought to reduce cost in bio-chemical technologies. Heterogeneous catalysts are simply recovered, reused and good for a large scale production compared to homogeneous catalysts. Nowadays, development of high efficiency heterogeneous catalysts for biomass conversion is one of challenges in this bio-fuel and bio-chemical industries.
Nanocatalysis and Molecular Simulation Research Group (NCAS) at NANOTEC mostly focuses on development and design of nanomaterials for energy and environmental applications. In the SIMulation research team, We have explored many heterogeneous catalytic reactions in bio-refinery processes including biomass conversion to biofuels/biochemicals and fuel improvement. The collaboration between simulation and experiment provides the deep understanding that benefits the bio-chemical/bio-fuel research area.
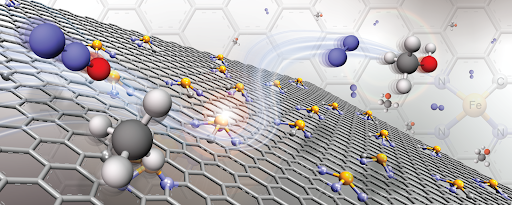
CATALYSIS AND ADSORBENT FOR POLLUTANT REMOVAL
Understanding insight into the molecular level of chemical processes in nanomaterials is a key of development of nanomaterials for specific purposes. Generally, the development of nanomaterials is essential to address challenges related to novel technologies including environmental applications. However, the design, development and commercialization of a new material can take several years. To accelerate functional material development, molecular simulation is a powerful tool that can help explore the nanomaterial at a significantly faster rate and lower cost than it can typically be done experimentally. This helps experimentalists focus only on the most promising materials. In this topic, deep understanding of the adsorption and reaction phenomenon of toxic compounds (such as Hg, NOx, H2S, etc) on nanomaterials at nanoscale are elucidated by molecular modelling and simulations. Through our computational design and calculations, promising materials were suggested before they have been synthesized and tested by experimentalists.
MATERIALS FOR ENERGY DEVICES
Our research applies atomistic simulation to elucidate the fundamental aspects of material properties and the underlying physical and chemical phenomena. The goal is to develop high-performance and low cost materials for
- Dye-sensitized solar cell and Organic light-emitting diode
- Metal-ion batteries
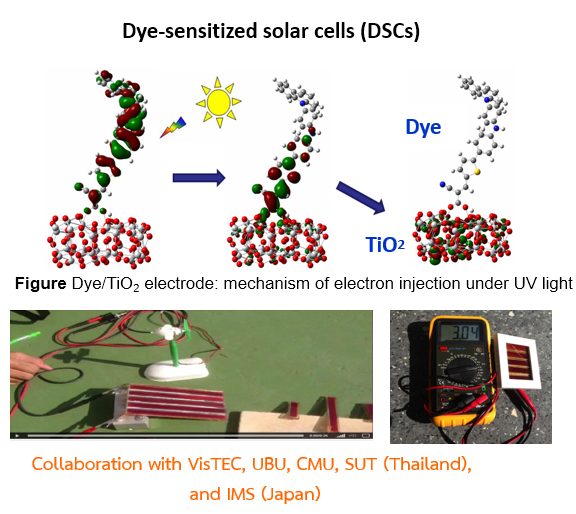
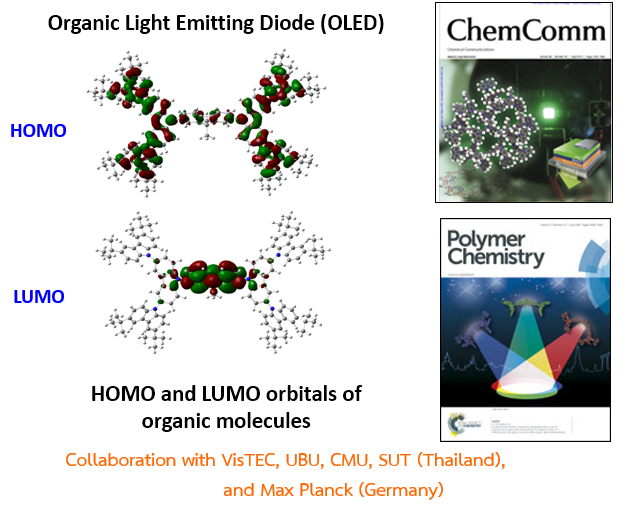
Dye-sensitized solar cell and organic light-emitting diode
The molecular geometry, electronic structure, and excited states of the organic molecules are very important for the design of new sensitizers for DSCs and OLEDs. We investigated these properties with density functional theory, time-dependent density functional theory, and the symmetry-adapted cluster-configuration interaction method to explain the behaviors in experiment. The double donor moieties of organic sensitizers not only contribute to enhancement of the electron-donating ability, but also inhibit aggregation between molecules and prevent iodide/triiodide in the electrolyte from recombining with injected electrons in the recombining with injected electrons in the semiconductor. In DSCs, our experimental and theoretical findings support the potential of direct electron injection from dye to TiO2 in one step with electronic excitation. The direct electron injection, inhibited aggregation, and high molar extinction coefficient may be the origin of the observed high efficiency in our experiment.

Metal-ion batteries
Metal-ion Batteries. In metal-ion batteries such as Li-ion, Na-ion, Li-S batteries, the solid electrolyte interphase (SEI) is formed on electrode surfaces from decomposition products of electrolytes. The SEI allows metal ions transport and blocks electrons in order to prevent further electrolyte decomposition and ensure continued electrochemical reactions. SEI properties strongly influence battery capacity and performance. The formation and growth mechanism of the nanometer thick SEI films are yet to be completely understood owing to their complex structure and lack of reliable in situ experimental techniques. Our research applies molecular simulation to investigate SEI on electrodes covering the thermodynamics and kinetics of electrolyte reduction reactions and SEI formation mechanisms in order to provide a better understanding of the complex SEI layer for the development of a highly efficient battery.